It is a rare and severe neurodegenerative disorder.
It belongs to a group of human and animal diseases known as transmissible spongiform encephalopathies (TSEs).
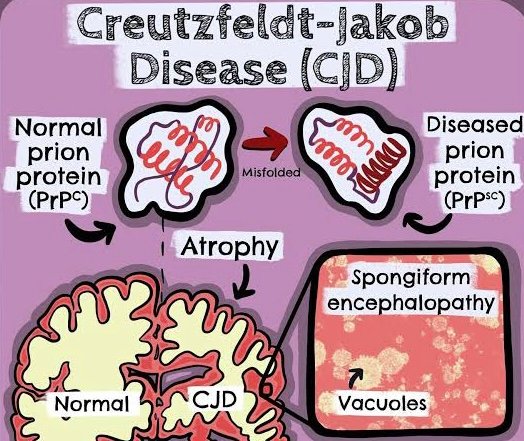
It is a PRION DISEASE
Unique features of this disease is that it can be transmitted as
INFECTIOUS
CONGENITAL
DEGENERATIVE DISORDER
1. Types:
- Sporadic CJD: Most cases occur spontaneously with no known cause.
- Familial CJD: Results from a genetic mutation. 10-12% are familial.
The gene for the prion protein is on Chromosome 20 - Acquired CJD: Linked to exposure to infected tissues or contaminated medical equipment.
2. Symptoms:
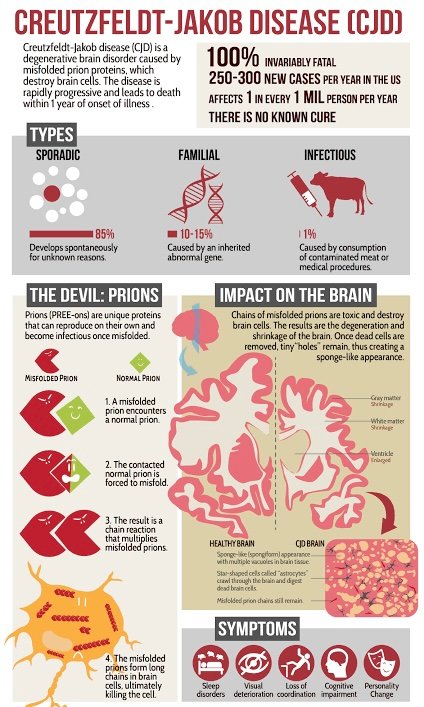
- Rapid Cognitive Decline: Memory loss(DEMENTIA), personality changes, impaired thinking, cerebellum Ataxia, Myoclonic Jerk, Akinetic mutism.
- Neuromuscular Issues: Muscle stiffness, twitching, weakness, coordination problems.
- Behavioral Changes: Depression, anxiety, irritability.
3. Progression:
- Symptoms progress rapidly over weeks to months.
- Severity varies, but deterioration is relentless.
- Most individuals succumb to the disease within a year of onset.
4. Diagnosis:
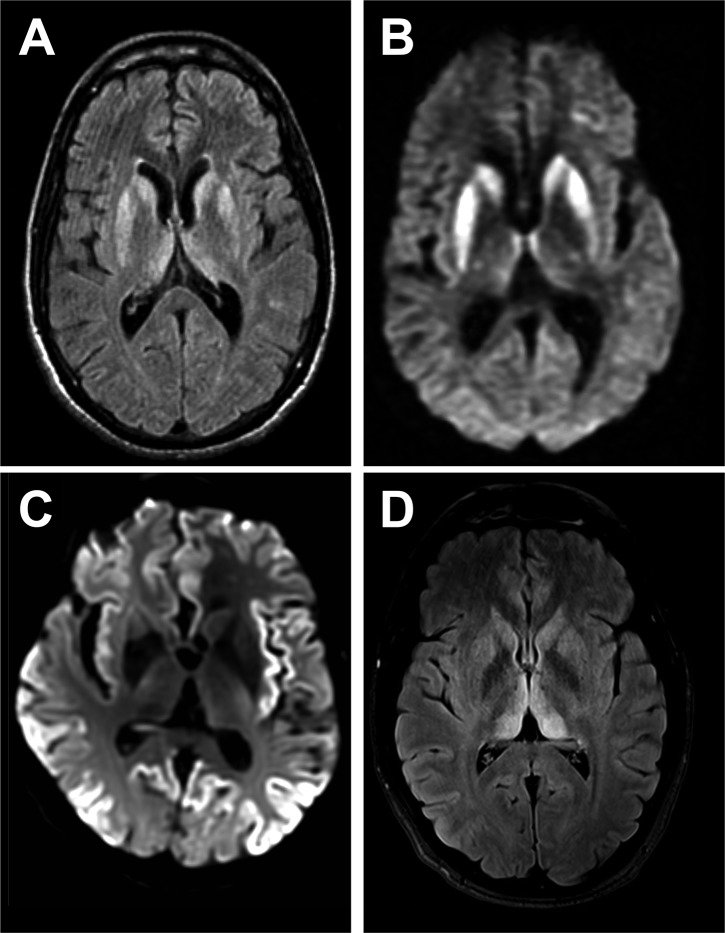
- Clinical evaluation of symptoms.
- Brain imaging (MRI, CT scans) to detect characteristic changes.
- Definitive diagnosis often involves a brain biopsy, but it’s rarely performed due to associated risks.
5. Causes:
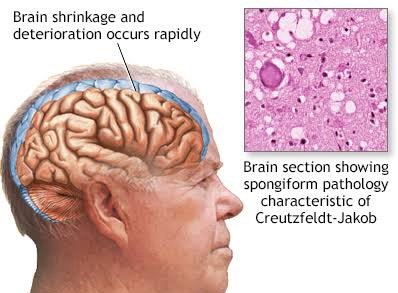
- Abnormal prion proteins accumulate in the brain, leading to neuronal damage.
- Prions are misfolded proteins that can induce normal proteins to misfold, spreading the disease.
- It can be transmitted through corneal transplantation
6. Treatment:
- No cure exists for CJD.
- Supportive care focuses on relieving symptoms and enhancing quality of life.
7. Impact:
- Its rapid progression and lack of effective treatment pose significant challenges.
- As there is LACK OF INFLAMMATION therefore there are likely to go in degenerative phase
8. Research and Awareness:
- Ongoing research aims to understand prion diseases and develop potential treatments.
- Raising awareness about CJD is crucial for early recognition and support.


{kind=link}